Critical points
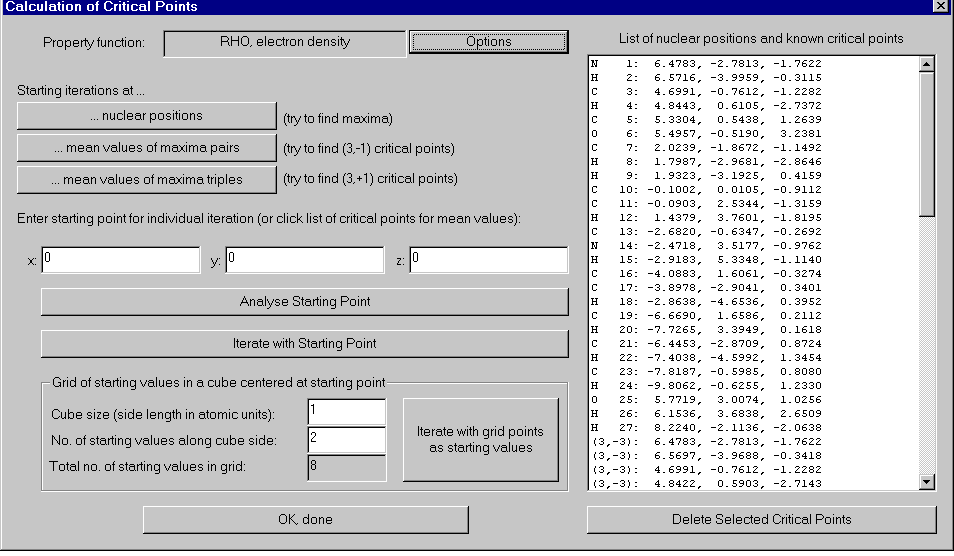
Critical points of the charge density are usually the first items you will want to
compute, since the whole theory of atoms in molecules is based on their properties. Of
course, critical points of other density functions can also be computed (see Control View).
At critical points all components of the gradient of the density function vanish. Apart
from the gradient, the Hessian matrix of second derivatives is also available at each
point (it is computed directly from the wave function or numerically).
Hence Newton's method can be employed to calculate the zeros of the gradient. Newton's
method turned out to be superior to all other methods like conjugate gradient or
quasi-Newton-methods. But it is heavily dependent on the choice of starting values.
AIM2000 helps you in several ways to choose starting values for a successful iteration:
- Most densities have maxima at or near the nuclear positions
of the atoms in the molecule. It is therefore sensible to use nuclear positions as
starting values. These starting values are tried when the button Starting
iterations at nuclear positions is clicked.
- Bonds in a molecule are represented by critical points of
the charge density with inertia (3, -1) which are located between two atoms. Hence the
mean value of two charge density maxima is usually a good starting value. These starting
values are tried when the button Starting iterations at mean values of maxima
pairs is clicked. Of course, the procedure can also be used for other densities.
- Ring-critical points are often found when starting with the
mean value of three charge density maxima. These starting values are tried when the button
Starting iterations at mean values of maxima triples is clicked. Of
course this procedure also works for others than the charge density. Caution! For
molecules with many atoms this procedure may be slow!
- If none of the above procedures finds the critical point you
suspect in a certain region, you can input a starting value of your own. This value can be
analyzed via the analyze-button and iteration started via the Iterate with
starting point-button.
You can also compose your starting values by clicking into the list of nuclear positions
and computed critical points. The composed starting point is the arithmetic mean of all
highlighted entries in the list.
- Some density functions (e.g. The Laplacian density)
frequently have a very high number of critical points. In order to find all critical
points in a certain region, you can define a grid of starting points in a cubic region of
space. The center of the cube is the preset starting point. You can set the length of a
cube-side and the number of starting values along the cube-side. Notice the total number
of starting points you defined! Iterate with grid points as starting values
starts the iteration.
For all types of starting point choices, the program will present a record of the
iterations and their success. Successfully computed critical points will be entered into
the right hand side list together with their inertia.
The Options-button brings you to a dialog where you can
manipulate the used Newton iteration process.
Delete selected critical points removes the highlighted critical
points from the list.
OK, done closes the dialog.